
Rilpivirine, tablet (film-coated), 25 mg (as hydrochloride), Edurant® - November 2011
Page last updated: 05 April 2012
Public Summary Document
Product: Rilpivirine, tablet (film-coated), 25 mg
(as hydrochloride), Edurant®
Sponsor: Janssen-Cilag Pty Ltd
Date of PBAC Consideration: November 2011
1. Purpose of Application
The submission sought a Section 100 (Highly Specialised Drugs
Program) Authority Required listing for the initial and continuing
treatment of Human Immunodeficiency Virus (HIV) infection in
combination with other antiretroviral agents in a patient with a
CD4 count of less than 500 per cubic millimetre or symptomatic HIV
disease.
Highly Specialised Drugs are medicines for the treatment of chronic
conditions, which, because of their clinical use or other special
features, are restricted to supply to public and private hospitals
having access to appropriate specialist facilities.
2. Background
This drug had not previously been considered by the PBAC.
3. Registration Status
Rilpivirine tablets were TGA registered on 23 December 2011 for the
indication:
In combination with other antiretroviral medicinal products, for
the treatment of human immunodeficiency virus type 1 (HIV-1)
infection in antiretroviral treatment naïve adult patients
with viral load less than or equal to 100,000 copies/mL at
baseline. This indication is based on Week 48 safety and efficacy
analyses from two randomised double blind, controlled Phase III
trials in treatment naïve adult patients and on Week 96 safety
and efficacy analyses from the Phase IIb trial TMC278-C204 in
treatment naïve adult patients.
4. Listing Requested and PBAC’s View
Section 100
Public Hospital Authority Required
(STREAMLINED)
Private Hospital Authority Required
Initial treatment of HIV infection in combination with other
antiretroviral agents in a patient with a CD4 count of less than
500 per cubic millimetre or symptomatic HIV disease;
Continuing treatment of HIV infection in combination with other
antiretroviral agents where the patient has previously received
PBS-subsidised therapy for HIV infection.
For PBAC’s view, see Recommendation and
Reasons.
5. Clinical Place for the Proposed Therapy
Human Immunodeficiency Virus (HIV) infection is a chronic,
immunosuppressive infection that is characterised by a continuous,
high-level viral replication and a slow, insidious, progressive
destruction of the human immune system. In the absence of effective
antiretroviral treatment, HIV infection leads to severe immune
deficiency and the development of the systemic opportunistic
infections and cancers that define the onset of the final stage of
HIV infection, the acquired immune deficiency syndrome (AIDS), and
ultimately results in death.
Typically, standard medical management of HIV-1 infection consists
of combinations of different antiretroviral therapies (e.g.
nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs),
non-nucleoside reverse transcriptase inhibitors (NNRTIs) and
protease inhibitors (PIs)).
The submission proposed rilpivirine as an alternative first-line
NNRTI therapy to efavirenz and nevirapine for treatment of
antiretroviral naïve HIV patients.
6. Comparator
The submission nominated efavirenz as the comparator which was
accepted by the PBAC.
7. Clinical Trials
The submission presented three randomised, double-blind, double-dummy trials and a
pooled analysis of two of the included trials comparing rilpivirine with efavirenz
in patients with treatment naïve HIV.
The ECHO and THRIVE trials were two Phase 3, randomised, controlled, non-inferiority
trials of 96 weeks duration, with a primary outcome of the proportion of patients
with plasma viral load less than 50 HIV-1 RNA copies/mL at Week 48 using the time
to loss of virologic response (TLOVR) algorithm. Both the ECHO and THRIVE trials used
a maximum allowable difference of 12% according to the TLOVR algorithm, defined by
the FDA as the non-inferiority criterion.
Trial C204 was a dose-finding trial, comparing three doses of rilpivirine blinded
(i.e. 25 mg, 75 mg or 150 mg once daily), to efavirenz 600 mg given open-label. The
primary outcome of C204 was the proportion of patients with plasma viral load less
than 50 HIV-1 RNA copies/mL at week 48 using the TLOVR algorithm.
Details of the studies published at the time of submission are shown in the following table:
| Trial ID / First author | Protocol title / Publication title | Publication citation | ||
|---|---|---|---|---|
| Direct randomised trials | ||||
| ECHO (C209) | A Phase III, randomized, double-blind trial of TMC278 25 mg q.d. versus efavirenz 600 mg q.d. in combination with a fixed background regimen consisting of tenofovir disoproxil fumarate and emtricitabine in antiretroviral-naïve HIV-1 infected subjects. |
Primary analysis (Week 48) Report date 28 May 2010 |
||
| Molina J-M et al 2011 | Rilpivirine versus efavirenz with tenofovir and emtricitabine in treatment naïve adults infected with HIV-1 (ECHO): a phase 3 randomised double-blind active controlled trial. | Lancet 2011: 378:238-246. | ||
| THRIVE (C215) | A Phase III, randomized, double-blind trial of TMC278 25 mg q.d. versus efavirenz 600 mg q.d. in combination with a background regimen containing 2 nucleoside/nucleotide reverse transcriptase inhibitors in antiretroviral-naïve HIV-1 infected subjects. |
Primary analysis (Week 48) Report date 08 June 2010 |
||
| Cohen CJ et al | Rilpivirine versus efavirenz with two background nucleoside or nucleotide reverse transcriptase inhibitors in treatment naïve adults infected with HIV-1 (THRIVE): a phase 2 randomised, non-inferiority trial. | Lancet 2011: 378:229-237 | ||
|
Pooled ECHO and THRIVE |
Pooling of two Phase III randomized, double-blind trials of TMC278 25 mg q.d. versus efavirenz 600 mg q.d. in combination with a background regimen containing 2 nucleoside/nucleotide reverse transcriptase inhibitors in antiretroviral-naïve HIV-1 infected subjects. |
Primary analysis (Week 48) Summary of Clinical Efficacy, 30 June 2010 Summary of Clinical Safety, 28 June 2010 Follow up analysis (Week 96) Report date 16 March 2011 Quality of life Report date 14 June 2010 |
||
| C204 | A Phase IIb randomized, partially blinded, dose-finding trial of TMC278 in antiretroviral naïve HIV-1 infected subjects. | Primary analysis (Week 48) Report date 07 June 2007 Follow up analysis (Week 96) Report date 12 March 2009 | ||
| Pozniak A.L. et al 2010 | Efficacy and safety of TMC278 in antiretroviral-naive HIV-1 patients: Week 96 results of a phase IIb randomized trial. | AIDS 2010; 24(1): 55-65. | ||
| Wilkin A. et al 2010 | TMC278 shows favourable tolerability and non-inferior efficacy compared to efavirenz over 192 weeks in HIV-1-infected treatment-naive patients. | Infection 2010; 38: S63-64. | ||
TMC278= rilpivirine
8. Results of Trials
The results for the primary outcome of the clinical trials, proportion of virological
responders, defined as a viral load less than 50 copies/mL at week 48 and 96 show
that both rilpivirine and efavirenz treatment resulted in high virologic response
rates (84.3% and 82.3% respectively in the pooled analysis).
Proportion of virologic responders (<50 HIV-1 RNA copies/mL, TLOVR), ITT
| Rilpivirine n/N (%) | Efavirenz n/N (%) | % Difference (95% CI) a | ||
|---|---|---|---|---|
| unadjusted | adjusted b | |||
| Week 48 | ||||
| ECHO | 287/346 (82.9%) | 285/344 (82.8%) | 0.1 (-5.5, 5.7) | -0.4 (-5.9, 5.2) |
| THRIVE | 291/340 (85.6%) | 276/338 (81.7%) | 3.9 (-1.6, 9.5) | 3.5 (-1.7, 8.8) |
| Pooled c | 578/686 (84.3%) | 561/682 (82.3%) | 2.0 (-2.0, 6.0) | 1.6 (-2.2, 5.3) |
| C204 | 74/93 (79.6%) | 72/89 (80.9%) | NR | -0.9 (-11.9, 10.1) |
| Week 96 | ||||
| ECHO | 263/346 (76.0%) | 271/344 (78.8%) | NR | -3.2 (-9.4, 3.1) |
| THRIVE | 269/340 (79.1%) | 258/338 (76.3%) | NR | 2.4 (-3.6, 8.4) |
| Pooled c | 532/686 (77.6%) | 529/682 (77.6%) | 0.0 (-4.4, 4.4) | -0.4 (-4.6, 3.8) |
| C204 | 71/93 (76.3%) | 63/89 (70.8%) | NR | 5.8 (-6.9, 18.5) |
NR = not reported; CI = confidence interval; ITT = intention to treat; TLOVR = time
to loss of virologic response
a For ECHO and THRIVE studies, CIs were calculated using the normal approximation
of the binomial distribution method.
b Difference in response rates predicted by logistic regression model adjusted for
factors including treatment, baseline viral load (continuous variable), background
regimen (for trial THRIVE and the pooled analysis only) and trial (for pooled analysis
only).
c The pooled analysis was for the ECHO and THRIVE trial.
The results for the proportions of virologic responders (<50 copies/mL, ITT TLOVR)
at Week 48 and Week 96 in the pooled analysis of ECHO and THRIVE by baseline viral
load category are presented in the table below. The proportion of virologic responders
decreased with increasing baseline viral load category in both treatment groups in
the pooled analysis. However, the proportion of virologic responders in the rilpivirine
group was greater than efavirenz for the ? 100,000 copies/mL category (Week 48: 90.2%
vs. 83.6%; Week 96: 84.0% vs. 79.9%) and lower than efavirenz for the > 500,000 copies/mL
category (Week 48: 69.6% vs. 75.6%; Week 96: 65.2% vs. 73.5%).
Proportion of Virologic Responders (< 50 HIV-1 RNA Copies/mL, TLOVR) at Week 48 and
96 by Baseline Viral Load Category (ECHO and THRIVE Pooled Analysis)
| Baseline viral load category (copies/?L) | Week 48 | Week 96 | ||
|---|---|---|---|---|
| RPV | EFV | RPV | EFV | |
| ?100,000 | 332/368 (90.2%) | 276/330 (83.6%) | 309/368 (84.0%) | 263/329 (79.9%) |
| >100,000 - ?500,000 | 198/249 (79.5%) | 223/270 (82.6%) | 178/249 (71.5%) | 205/270 (75.9%) |
| >500,000 | 48/69 (69.6%) | 62/82 (75.6%) | 45/69 (65.2%) | 61/83 (73.5%) |
Abbreviations:EFV = efavirenz; HIV = human immunodeficiency virus; RNA = ribonucleic
acid;
RPV = rilpivirine; TLOVR = Time to loss of virologic response.
The results for differences between rilpivirine and efavirenz (95% CI) in response
rates at week 48 for the ECHO and THRIVE intention to treat (ITT) populations showed
that the pre-defined non-inferiority criteria in both trials were both met as the
lower limit of the 95% confidence intervals were above -12% (the maximum allowable
difference according to the time to loss of virologic response (TLOVR) algorithm as
defined by the FDA).
Differences between RPV and EFV (95% CI) in response rates at week 48 for ECHO and THRIVE (ITT)
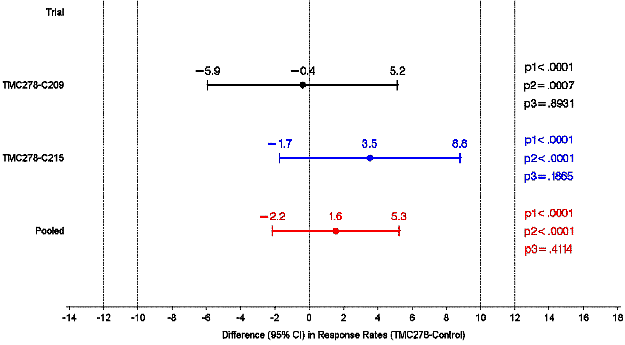
CI = confidence interval; EFV = efavirenz; HIV = human immunodeficiency virus; RPV
= rilpivirine; TMC278 = rilpivirine.
Difference in response rates predicted by logistic regression model including factors
treatment, baseline viral load (continuous variable), background regimen (for THRIVE
and the pooled analysis only) and trial (for pooled analysis only). p1 = non-inferiority
at 12% margin, p2 = non-inferiority at 10% margin, p3 = p-value for superiority.
In the 48 week pooled snapshot analysis of the ECHO and THRIVE analyses, 12.8% of
the rilpivirine patients and 9.1% of the efavirenz patients had virologic failures
(risk difference 3.7%; 95% CI 0.4% to 7.0%). In these groups, more patients treated
with rilpivirine discontinued treatment due to virologic failure than patients treated
with efavirenz (4.7% vs. 1.8%, respectively; risk difference 2.9%; 95% CI: 1.0 to
4.8%).
Subjects participating in ECHO and THRIVE were included in the exploratory resistance
analyses if they experienced virologic failure. Similar proportions of virologic failures
developed treatment emergent RT resistance-associated mutations (RAMs) (Week 96: rilpivirine
= 69/86 (80%); efavirenz = 36/42 (86%)). However, virologic failures with phenotypic
resistance to rilpivirine generally exhibited more than 1 mutation (typically E138K
and M184I), resulting in cross-resistance to efavirenz, etravirine and to a lesser
extent nevirapine. Virologic failures with phenotypic resistance to efavirenz were
typically mutations (K103N and M184V), and therefore demonstrated high cross-resistance
to nevirapine, but retained susceptibility to etravirine and rilpivirine.
In the 96 Week analysis 35 (43.2%) of 81 rilpivirine virologic failures with resistance
data had phenotypic resistance to rilpivirine at failure. Among the 35 failures resistant
to rilpivirine, 30 (85.7%), 32 (91.4%) and 16 (45.7%) were cross-resistant to efavirenz,
etravirine and nevirapine, respectively. Seventeen (41.5%) of 41 efavirenz virologic
failures had phenotypic resistance to efavirenz at failure. Among the 17 failures
resistant to efavirenz none, one (5.9%) and 15 (88.2%) were cross-resistant to rilpivirine,
etravirine and nevirapine, respectively.
Resistance to emtricitabine and lamivudine was observed in the majority of rilpivirine
virologic failures, and to a lesser degree in the efavirenz virologic failures. More
than 90% of the rilpivirine and efavirenz virologic failures retained susceptibility
to tenofovir, abacavir, and zidovudine at failure.
For PBAC’s view of these results, see Recommendations and Reasons.
A summary of the adverse events reported in the pooled ECHO and THRIVE trials at 96
weeks showed that there were no differences in the number of adverse events (AEs)
and serious adverse events (SAEs) between rilpivirine and efavirenz at week 96. However,
more efavirenz treated patients permanently discontinued treatment due to an adverse
event than rilpivirine treated patients (8.5% versus 4.1%; RR 0.48 [95% CI: 0.31,
0.74]).
Patients treated with rilpivirine were less likely to have any skin event (rash),
neuropsychiatric event, or neurologic event than patients treated with efavirenz.
9. Clinical Claim
The submission claimed that rilpivirine is non-inferior to
efavirenz in terms of efficacy and safety.
The PBAC considered the submission’s claim that rilpivirine
is non-inferior to efavirenz in terms of efficacy and safety was
reasonable, but noted that rilpivirine is associated with more
virological failure and efavirenz is associated with more treatment
related adverse events.
10. Economic Analysis
The submission presented a cost minimisation analysis. The PBAC
agreed that the equi-effective doses are rilpivirine 25 mg once
daily over 96 weeks and efavirenz 600 mg once daily over 96 weeks,
based on the fixed dosing used in the ECHO and THRIVE trials.
11. Estimated PBS Usage and Financial Implications
The submission estimated the likely number of patients treated to
be less than 10,000 per year by Year 5. The estimate is uncertain
based on the possible uptake rate and whether rilpivirine will
substitute for other antiretroviral agents.
The net financial cost/year to the PBS was estimated by the
submission to be less than $10 million over the first 5 years. The
estimate is uncertain, due to uncertainty as to whether rilpivirine
will replace other antiretroviral agents that have a different
price compared to the proposed rilpivirine price.
12. Recommendation and Reasons
The PBAC recommended the listing of rilpivirine tablets on the PBS
in the Section 100 Highly Specialised Drugs Program as Private
Hospital Authority Required and Public Hospital Authority Required
(Streamlined) listings for the initial and continuing treatment of
HIV infection in combination with other anti-retroviral agents in a
patient with a CD4 count of less than 500 per cubic millimetre or
symptomatic HIV disease on a cost-minimisation basis with
efavirenz. The equi-effective doses are rilpivirine 25 mg once
daily over 96 weeks and efavirenz 600 mg once daily over 96 weeks,
based on the fixed dosing used in the ECHO and THRIVE trials.
The PBAC accepted that efavirenz is the appropriate
comparator.
The PBAC noted that the TGA Delegate’s Summary had been
received just prior to the PBAC meeting and that the Delegate
proposed to approve rilpivirine for registration for the treatment
of HIV-1 infection in anti-retroviral treatment-naïve adult
patients in combination with other anti-retroviral medicinal
products. Therefore, the PBAC considered that although there was
some uncertainty about the place of rilpivirine therapy in practice
as the data presented were from treatment naïve patients only,
the requested restriction did not prevent the use of rilpivirine in
patients who could not tolerate another first-line therapy.
The PBAC noted the cross-resistance of rilpivirine to other
non-nucleoside reverse transcriptase inhibitors (NNRTIs) and
nucleoside/tide reverse transcriptase inhibitors (N(t)RTIs), and
that this may have an impact on the choice of NNRTI for first-line
treatment. However, the PBAC was confident that prescribers
treating patients with HIV had the necessary expertise that would
ensure the appropriate use of rilpivirine. Therefore, the PBAC
considered that a restriction wording consistent with that of the
comparator efavirenz was appropriate.
The PBAC considered that the clinical trial data presented from the
ECHO and THRIVE trials demonstrates the non-inferiority of
rilpivirine compared with efavirenz for the primary outcome of the
proportion of patients with a plasma viral load of less than 50
copies per mL at week 48. Both rilpivirine and efavirenz treatment
resulted in high virologic response rates (84.3% and 82.3%
respectively in the pooled analysis) and the pre-defined
non-inferiority criteria in both trials were both met as the lower
limit of the 95% confidence intervals were above -12% (the maximum
allowable difference according to the time to loss of virologic
response (TLOVR) algorithm as defined by the FDA).
The PBAC noted that patients treated with rilpivirine were more
likely to have treatment failure due to virologic failure (all
patients and for patients with a viral load of >100,000
copies/mL at baseline), while patients treated with efavirenz were
more likely to have treatment failure due to discontinuation due to
adverse events. The differences in virologic failure were more
pronounced for patients with a higher virological load at
baseline.
The majority of rilpivirine virologic failures resistant to
rilpivirine were cross-resistant to efavirenz (85.7%) and
etravirine (91.4%), while approximately half were cross-resistant
to nevirapine (45.7%). Patients with efavirenz virologic failure
resistant to efavirenz were mostly cross-resistant to nevirapine
(88.2%), while no patients were cross-resistant to rilpivirine
(0%).
From the pooled results of the trials, the PBAC noted that there
were no differences in the number of adverse events (AEs) and
serious adverse events (SAEs) between rilpivirine and efavirenz at
week 96. However, more efavirenz treated patients permanently
discontinued treatment due to an adverse event than rilpivirine
treated patients (8.5% versus 4.1%; RR 0.48 [95% CI: 0.31,
0.74]).
Therefore, the PBAC considered the submission’s claim that
rilpivirine is non-inferior to efavirenz in terms of efficacy and
safety was reasonable, however, noting that rilpivirine is
associated with more virological failure and efavirenz is
associated with more treatment related adverse events that lead to
discontinuation.
The PBAC considered that the listing of rilpivirine would provide
an alternative treatment option to currently listed NNRTIs and
would be unlikely to increase the cost to the PBS of treating
patients with HIV.
The PBAC noted the advice of the Highly Specialised Drugs Working
Party which supported listing rilpivirine as a HSD under Section
100.
Recommendation:
RILPIVIRINE, tablet (film-coated), 25 mg (as hydrochloride)
Restriction: Section 100
Public Hospital Authority Required (STREAMLINED)
Private Hospital Authority Required
Initial treatment of HIV infection in combination with other antiretroviral agents in a patient with a CD4 count of less than 500 per cubic millimetre or symptomatic HIV disease;
Continuing treatment of HIV infection in combination with other antiretroviral agents where the patient has previously received PBS-subsidised therapy for HIV infection.
Maximum qty: 60
Rpt: 5
13. Context for Decision
The PBAC helps decide whether and, if so, how medicines should be
subsidised in Australia. It considers submissions in this context.
A PBAC decision not to recommend listing or not to recommend
changing a listing does not represent a final PBAC view about the
merits of the medicine. A company can resubmit to the PBAC or seek
independent review of the PBAC decision.
14. Sponsor’s Comment
The sponsor had no further comment.
