
Aflibercept, 2 mg/0.05 mL injection, 1 x 0.05 mL vial and 2 mg/0.05 mL injection, 1 x 0.05 mL syringe, Eylea® - March 2014
PDF printable version of this page
Public Summary Document
Product: Aflibercept, 2 mg/0.05 mL injection, 1 x 0.05 mL vial and 2 mg/0.05 mL injection,
1 x 0.05 mL syringe, Eylea®
Sponsor: Bayer Australia Ltd
Date of PBAC Consideration: March 2014
1. Purpose of Application
To seek an Authority required listing for aflibercept for treatment of patients with macular oedema secondary to central retinal vein occlusion (CRVO).
2. Background
This was the second submission for aflibercept considered by the PBAC that sought to extend the listing of aflibercept to include the treatment of visual impairment due to macular oedema secondary to CRVO.
At its July 2013 meeting, the PBAC rejected the initial submission for aflibercept in this condition on the basis of an unacceptably high and likely underestimated incremental cost effectiveness ratio (ICER) compared with best supportive care, and on the basis of inadequate comparative data against either bevacizumab or ranibizumab.
The PBAC advised that a resubmission would need to provide comparative efficacy data against bevacizumab and a more formal comparison against ranibizumab, because the PBAC considered that these two drugs are currently being used in practice and would be likely to be replaced by aflibercept if it were PBS listed.
The initial submission was a co-dependent submission that included the use of OCT. The resubmission assumed that the place of OCT in the treatment of CRVO would be determined by parallel MSAC processes that were underway.
3. Registration Status
As of 1 May 2014, aflibercept is TGA registered in adults for the treatment of:
- neovascular (wet) age-related macular degeneration (wet AMD)
- visual impairment due to macular oedema secondary to central retinal vein occlusion (CRVO).
4. Listing Requested and PBAC’s View
Authority required
Initial treatment, by an ophthalmologist, of visual impairment due to macular oedema secondary to central retinal vein occlusion (CRVO) in patients with:
a) a confirmed presence of central retinal thickening on OCT
b) presence of documented impairment of BCVA on the ETDRS chart
Initial treatment lasts for up to six four-weekly injections and is followed by OCT guided treatment.
Authority required
Continuing treatment by an ophthalmologist for macular oedema secondary to CRVO where the patient has previously been granted an authority prescription for the same eye.
The requested restriction was unchanged from the July 2013 submission and included a requirement that patients have confirmed central retinal thickening on optical coherence tomography (OCT) and that continuing treatment be guided by OCT.
Listing was requested on the basis of a cost-utility analysis versus placebo as a proxy for best supportive care. A cost-minimisation analysis versus ranibizumab was also presented in the resubmission.
5. Clinical Place for the Proposed Therapy
Retinal vein occlusion is an obstruction of the veins draining blood from the back of the eye, caused by a blood clot (thrombus) or other possible causes such as external compression of the vein or diseases of the vessel wall. Obstruction of one or more of the retinal veins causes blood and fluid to leak from the capillaries that drain into the obstructed vein. This can result in swelling or thickening of the retina (oedema). It is classified into either branch retinal vein occlusion (BRVO) or CRVO based on the site of venous occlusion. The condition is predominantly unilateral (i.e. affects one eye only).
The resubmission proposed aflibercept as first-line therapy for macular oedema caused by CRVO. This was the same clinical place as proposed in the previous submission.
In its July 2013 consideration of aflibercept for CRVO, the PBAC noted that there is unsubsidised use of ranibizumab (approved by TGA) and bevacizumab (not approved by TGA for this indication).
6. Comparator
The PBAC considered that best supportive care in the absence of injected therapy was a relevant comparator and that the submitted sham-controlled trials provided relevant information for PBAC to compare aflibercept with best supportive care.
The PBAC considered that compounded bevacizumab was also a relevant comparator because evidence of its current use in patients who would be eligible for PBS-subsidised aflibercept means that prescribers would replace compounded bevacizumab in practice with aflibercept if the PBS subsidises aflibercept.
The PBAC noted the applicant's arguments against accepting bevacizumab as a comparator, principally on the grounds that this represents use beyond the TGA-approved indications and adapting a formulation that is not intended for injection into the eye. However, the PBAC noted that Subsection A.4 of the PBAC Guidelines states that "PBAC bases its judgement about the main comparator on what would be likely to happen, rather than what should happen, in keeping with the above definition of the main comparator." The PBAC noted that an evidence base beyond the sham-controlled trials was needed to assess the comparison of compounded bevacizumab with aflibercept.
The PBAC noted Departmental advice that, if compounded bevacizumab were to be considered for PBS listing, its pricing for PBS purposes would most likely reflect the approach to pricing already applied to bevacizumab as an oncology medicine through the Efficient Chemotherapy Funding Program.
The PBAC noted that the resubmission included an indirect comparison with ranibizumab in the main body of the submission. This indirect comparison was presented in an appendix in the July 2013 submission.
7. Clinical Trials
The resubmission was based on two direct randomised trials, COPERNICUS and GALILEO, comparing aflibercept to placebo/sham injection. These were the same two trials presented in the July 2013 submission. These two trials are also included in the indirect comparison against ranibizumab, with the CRUISE randomised trial against a sham injection control providing ranibizumab data for patients with CRVO.
A summary of the trials is shown in the table below:
|
Trial ID/ first author |
Protocol title/ publication title |
Publication citation |
|---|---|---|
|
COPERNICUS |
||
|
Boyer, D et al. |
Vascular endothelial growth factor Trap-Eye for macular edema secondary to central retinal vein occlusion: six-month results of the phase 3 COPERNICUS study. |
Ophthalmology 2012; 119(5):1024-32. Epub March 21, 2012. |
|
Brown, DM et al. |
Intravitreal Aflibercept Injection for Macular Edema Secondary to Central Retinal Vein Occlusion: 1-Year Results From the Phase 3 COPERNICUS Study. |
American Journal of Ophthalmology 2012; article in press. |
|
Gillies, M |
Intravitreal VEGF trap-eye in central retinal vein occlusion: Results of the phase 3 Copernicus and Galileo studies. |
Clinical and Experimental Ophthalmology 2012; 40(S1):44
RANZCO 44th Annual Scientific Congress. |
|
GALILEO |
||
|
Holz, F G et al. |
VEGF Trap-Eye for macular oedema secondary to central retinal vein occlusion: 6-month results of the phase III GALILEO study. |
British Journal of Ophthalmology (2012) Epub ahead of print; January 7, 2012. |
|
Gillies, M |
Intravitreal VEGF trap-eye in central retinal vein occlusion: Results of the phase 3 Copernicus and Galileo studies. |
Clinical and Experimental Ophthalmology 2012; 40(S1):44
RANZCO 44th Annual Scientific Congress. |
|
CRUISE |
||
|
Brown, DM et al
Campochiaro, PA et al |
Ranibizumab for Macular Oedema following Central Retinal Vein Occlusion: Six-Month Primary End Point Results of a Phase III Study.
Sustained Benefits from Ranibizumab for Macular Oedema following Central Retinal Vein Occlusion: Twelve-Month Outcomes of a Phase III Study. |
Ophthalmology 2010; 117(6):1124-33
Ophthalmology 2011; 118(10):2041-9 |
The key features of the included trials are presented in the table below.
|
Trial |
N |
Design/ duration |
Risk of bias |
Patient population |
Outcome(s) |
Use in economic model |
|---|---|---|---|---|---|---|
|
Aflibercept versus sham injection |
||||||
|
COPERN-ICUS |
187 |
R, DB 100 wks |
Low |
Patients with ≥250µm CST as measured by OCT and an ETDRS BCVA of 20/40 to 20/320 |
Proportion of patients with ≥15 letter gain in BCVA at 24/52 |
Used |
|
GALILEO |
171 |
R, DB 76 wks |
Low |
Used |
||
|
Ranibizumab versus sham injection used for the indirect comparison |
||||||
|
CRUISE |
392 |
R, DB |
Low |
Patients with ≥250µm CST as measured by OCT and a Snellen equivalent BCVA of 20/40 to 20/320 |
Mean change from baseline in BCVA at Month 6 |
Not used |
Abbreviations: BCVA=best corrected visual acuity; CST=central subfield thickness; DB=double blind; ETDRS=Early Treatment Diabetic Retinopathy Study; OCT=optical coherence tomography; R=randomized. Source: compiled during the evaluation
The PBAC noted that an indirect comparison against bevacizumab was conducted during the evaluation. This indirect comparison involved the Epstein 2012 trial which compared bevacizumab and sham injection in 60 patients (Epstein DL, Algvere PV, von Wednt G, Seregard S, Kvanta A. Bevacizumab for macular edema in central retinal vein occlusion: a prospective randomized double masked clinical study. Ophthalmology 2012;119(6):1184-9).
Dose response
The PBAC noted that the proposed product information for aflibercept specified that treatment of CRVO is initiated with three loading doses given monthly (before continuing on a “treat and extend” regimen), whereas the trials presented in the resubmission used up to six loading doses given monthly. The PBAC noted that, after three to four doses, the dose-response curves from the GALILEO and COPERNICUS trials (Figures 1 and 2) indicated a relative flattening in response in terms of mean change in best corrected visual acuity (BCVA) (with the maximum response for this outcome being achieved at approximately 24 weeks).
Mean change from baseline to week 52 and week 100 in visual acuity by treatment group for the COPERNICUS trial
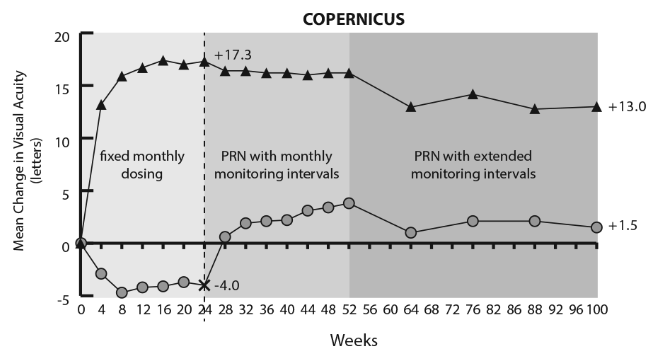
![]()
Mean change from baseline to week 52 and week 76 in visual acuity by treatment group for the GALILEO study
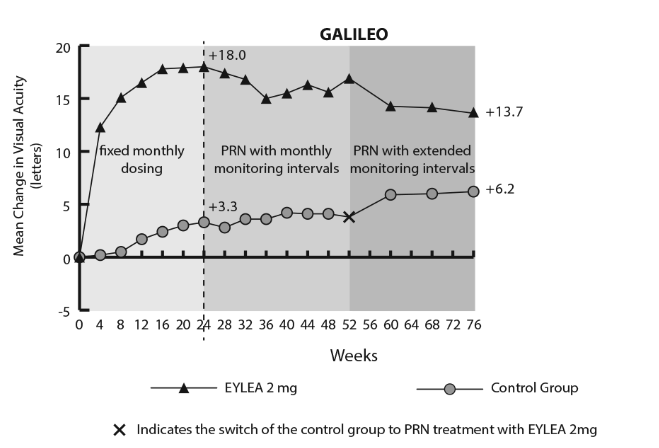
The PBAC noted that the resubmission did not provide any evidence relating the revised loading dose to clinical efficacy beyond three months. Further, the resubmission did not provide any evidence relating to whether this would affect the number and frequency of injections required as a result of using the “treat and extend” approach to managing aflibercept therapy.
The PBAC noted that no consumer comments were submitted in relation to this submission. The sponsor of this submission did not request a hearing.
8. Results of Trials
The results presented in the resubmission comparing aflibercept with sham injections were unchanged from those presented in the July 2013 submission. The results have been previously reported in the July 2013 Public Summary Document for aflibercept, available on the PBS website.
The PBAC recalled that in July 2013 it had accepted that, based on the results of the COPERNICUS and GALILEO trials and the primary outcome of BCVA, aflibercept is clinically and statistically significantly more efficacious than placebo.
However, the PBAC re-iterated its previous concerns regarding the long-term efficacy of aflibercept, which were:
- the PBAC was unable to assess the longer term treatment effect of aflibercept for patients who crossed-over (at Week 24 in COPERNICUS and Week 52 in GALILEO) as these data were not available
- there was deterioration in treatment effect following the initial 24 weeks when dosing was changed to an ‘as needed’ basis. The clinical trial report for COPERNICUS noted that there appeared to be recurrence of macular oedema when patients switched to ‘as needed’ dosing.
The PBAC recalled that in July 2013 it had noted the wide confidence intervals from the results of the indirect comparison against ranibizumab, but considered that on balance, aflibercept was likely to be no worse than ranibizumab in regards to comparative efficacy based on 24 week data.
However, the PBAC re-iterated its July 2013 advice that it could not draw any conclusions from the week 52 data, because sham-injection patients in COPERNICUS and CRUISE were able to cross-over to active treatment following week 24, and the 52 week results provided did not analyse patients according to the duration of treatment. Neither the original submission nor the resubmission provided results for patients who commenced active treatment after 24 weeks separately.
The results of the indirect comparison against bevacizumab, which was conducted during the evaluation, are presented below.
|
Trial ID |
Aflibercept trials |
Bevacizumab trials |
Indirect OR |
||||
|
OR (95% CI) |
Aflibercept n/N (%) |
Sham injection n/N (%) |
Sham injection n/N (%) |
Bevacizumab n/N (%) |
OR |
||
|
24 weeks – proportion of patients who gained ≥15 letters in BCVA |
|||||||
|
COPERN-ICUS |
9.10 (4.13, 20.05) |
64/114 (56.1%) |
9/73 (12.3%) |
|
|
|
|
|
GALILEO |
5.34 (2.66, 10.72) |
62/103 (60.2%) |
15/68 (22.1%) |
|
|
|
|
|
Pooled |
6.74 (4, 11.37) |
|
|
|
|
|
|
|
Epstein et al 2012 |
|
|
|
6/30 (20%) |
18/30 (60%) |
6.00 (1.89, 19.04) |
|
|
Indirect |
P=0.8573 |
1.12 (0.32, 3.99) |
|||||
Source: calculated during the evaluation
OR=odds ratio
The PBAC noted the risk of bias across the indirect comparison relating to the different study population characteristics, but noted that the results indicated non-inferiority between bevacizumab and aflibercept in terms of efficacy.
In relation to comparative harms, the PBAC recalled that the previous submission provided only grouped events (i.e. ‘treatment emergent’ or serious adverse events) and did not provide any information on specific events. The resubmission included specific adverse events.
The PBAC noted that the trial data showed minimal differences in adverse events between aflibercept and sham injection within the two aflibercept trials (COPERNICUS and GALILEO).
The PBAC noted that the resubmission provided updated post-marketing data based on the most recent United States periodic safety update report for aflibercept, and had identified no new evidence that would change the benefit-risk profile of aflibercept.
The PBAC noted that the indirect comparison between aflibercept and ranibizumab indicated that there was no statistically significant difference between the two drugs in terms of adverse events.
|
Adverse event |
Indirect analysis |
|
|
RD (95% CI) |
RR (95% CI) |
|
|
Intraocular inflammation |
RD=0 (-0.06, 0.06) |
RR=0.8 (0.05, 14.20) |
|
Cataract |
RD=0.03 (-0.01, 0.07) |
RR=9.8 (0.12, 789.22) |
|
Iris neovascularisation |
RD=0.05 (-0.50, 0.60) |
RR=4.36 (0.13, 146.90) |
|
Neovascular glaucoma |
RD=-0.01 (-0.04, 0.02) |
RR=0.65 (0.02, 24.27) |
|
Retinal tear |
RD=-0.01 (-0.05, 0.03) |
RR not estimable |
|
Vitreous haemorrhage |
RD=-0.01 (-0.11, 0.09) |
RR=0.15 (0.01, 1.89) |
|
Myocardial infarction |
RD=-0.01 (-0.05, 0.03) |
RR=0.22 (0.00, 14.58) |
|
Hypertension |
RD=0.01 (-0.04, 0.06) |
RR not estimable |
RD=risk difference; RR=relative risk
The PBAC noted that, because it had considered compounded bevacizumab to be a relevant comparator, a comprehensive review of the comparative effectiveness and safety of aflibercept and compounded bevacizumab would be informative. An analysis of the comparative safety would need to consider:
- injection site reactions, and
- harms associated with the systemic effects of vascular endothelial growth factor (VEGF) inhibition.
The incidence and severity of many of the harms would be unlikely to vary between ocular conditions, therefore the analysis would be best informed by safety data from all the trials of bevacizumab and aflibercept across all ocular indications (including AMD). This would increase the evidence base available for a comparison of safety. Similarly, if there is a class effect across anti-VEGF medicines, this would become clearer if consistent conclusions of non-inferiority and equi-effective doses are reached across ocular conditions.
A summary of the comparative benefits and harms for aflibercept versus placebo (sham injection) is presented in the table below.
|
Trial |
Aflibercept |
Sham injection |
RR* (95% CI) |
Event rate/100 patients |
RD* (95% CI) |
|
|
Aflibercept |
Sham injection |
|||||
|
Benefits |
||||||
|
Gain ≥15 letters in BCVA |
||||||
|
GAL & COPa |
126/217 |
24/141 |
3.37 (2.04, 5.57) |
58.1 |
17.0 |
0.41 (0.32, 0.50) |
|
Harms |
||||||
|
Intraocular pressure increased |
||||||
|
GAL & COPa |
17/218 |
9/142 |
1.22 (0.55, 2.67) |
7.8 |
6.3 |
0.01 (-0.04, 0.07) |
|
Hypertension |
||||||
|
GAL & COPa |
14/218 |
7/142 |
1.29 (0.53, 3.14) |
6.4 |
4.9 |
0.01 (-0.04, 0.06) |
* RR and RD calculated using Stats Direct Version 2.7.9; a pooled results of COPERNICUS and GALILEO trials;
Source: harms compiled from the resubmission
BCVA=best corrected visual acuity; GAL=GALILEO trial; COP=COPERNICUS; RR=relative risk; RD=risk difference.
The PBAC noted that, based on these trials, for every 100 patients treated with aflibercept compared to sham injection:
- approximately 41 extra patients would experience a gain of at least 15 letters in visual acuity (VA) (from baseline to 24 weeks)
- approximately one or two extra patients would experience an increase in intraocular pressure
- approximately one or two extra patients would experience an increase in intraocular pressure.
9. Clinical Claim
As in July 2013, the resubmission claimed that aflibercept has superior effectiveness with a similar safety profile compared to placebo. The PBAC accepted this claim in July 2013. The PBAC saw no reason to change its previous conclusion in its consideration of the resubmission.
Further, the PBAC considered that the three loading dose regimen recommended by the TGA would be likely to be used in practice rather than the six loading dose regimen used in the trials, however the extent of use of each regimen in clinical practice was unknown. The PBAC noted that the resubmission did not provide any evidence relating the revised loading dose to longer-term clinical efficacy.
As in July 2013, the resubmission claimed that aflibercept is non-inferior in terms of efficacy and safety compared to ranibizumab. The PBAC accepted this claim in July 2013 based on comparative data at week 24. The PBAC saw no reason to change its previous conclusion in its consideration of the resubmission.
The PBAC accepted that, on the basis of the analysis conducted during the evaluation, aflibercept is non-inferior in terms of efficacy compared to bevacizumab.
10. Economic Analysis
A modelled economic evaluation (cost-utility analysis) was presented based on the claim of superior effectiveness and similar safety of aflibercept compared to placebo. The resubmission estimated ICERs of between $45,000 and $75,000 per quality adjusted life year (QALY) with and without OCT costs included in the placebo arms.
The PBAC noted that the structure of the model remained unchanged from the previous submission, but the following inputs had changed:
- the age of onset had increased from 64 to 72 years, which the PBAC noted shortened the model duration from 36 to 28 years;
- the mortality risk of unilateral blindness had decreased from a relative risk of 1.23 to 1.13;
- OCT costs had decreased;
- the aflibercept price was revised.
The PBAC accepted the advice of the ESC that the overall patient impact of unilateral blindness has been shown to be modest. The PBAC noted that the utility values in the GALILEO trial showed that the utility decrement from near perfect vision in the affected eye to being blind in the affected eye is relatively modest (0.07 from BCVA ≥80 to BCVA <35; 0.931 vs. 0.858).
The PBAC noted that the pre-PBAC response stated that:
- the ‘health states are defined by the BCVA of the affected eye’
- the utility values employed by the model (i.e. EQ-5D values in GALILEO) reflect overall VA.
The PBAC further noted that the utility values from GALILEO are pooled across baseline, 24 weeks and 52 weeks, and while they relate to overall VA (both eyes), they are reported for 5 VA states allowing transitions for the CRVO-affected eye. Thus, the model overestimates the utility gains associated with transitions for the CRVO-affected eye to the extent that the mapping to utility values also implies transitions in the other (usually better seeing) eye.
The model structure relied on assuming a link between BCVA of the CRVO-affected eye (which forms the basis of the transition probabilities used) and QoL or utility values which are affected by the overall BCVA of the patient.
Therefore, the PBAC agreed with the ESC that the link between BCVA of the CRVO-affected eye and overall utility significantly biases the incremental cost-effectiveness over placebo in favour of aflibercept.
The PBAC noted that the ESC had requested data to enable an extensive respecification of the base case to account for this structural issue, given the paucity of data available to inform an alternative model structure. The PBAC noted that such data was not provided in the pre-PBAC response.
The PBAC noted that the premise underlying the ESC’s suggestions for possible alternative approaches was that a difference in utilities can only be attributed to those patients that experience a perceptible difference in overall VA and should reflect the extent of this perceptible difference in overall VA. The PBAC considered that these alternative approaches might be feasible to address this concern with the submitted model, including:
- adjusting for the proportion of patients for whom the CRVO-affected eye is the better or worse seeing eye at baseline and then using the current model’s differences across utilities for patients for whom the better seeing eye is affected and reduced differences across utilities for patients for whom the worse seeing eye is affected (because the better seeing eye accounts for most of the contribution of VA on overall utility)
- similarly, if the trials collected BCVA data for both eyes at one or more time points after baseline then it might be possible to estimate any difference across the arms in the proportions of patients for whom the CRVO-affected eye improves from being the worse seeing eye at baseline to the better seeing eye after treatment. If this improvement were sufficient to be discernible to the patient, this could become the basis of estimating a utility gain in such patients where the effect on the treated eye’s VA might have a direct and discernible effect on overall VA.
Alternatively, if the trials collected VA data for both eyes independently (at baseline and one or more time points after baseline), then it might be possible to incorporate the VA of both the better seeing eye and the worse seeing eye in the model’s health states. That is, each health state would incorporate two variables simultaneously: the visual acuity of the worse seeing eye and the visual acuity of the better seeing eye, and transitions across these health states would reflect the possible transitions for the CRVO-affected eye whilst holding the VA of the other eye constant. This would require utilities and transition probabilities to be available for each of these health states. The PBAC acknowledged, however, that there might be insufficient data available to adequately inform a greater number of health states.
The PBAC agreed with the ESC that, as overall VA is influenced mostly by the better seeing eye, the potential for utility differences to arise is influenced by the baseline proportions of worse and better seeing eyes that are affected with CRVO. Inclusion of only the proportions of patients for which overall VA differences would be perceptible for patients would enable the model to be based on patient-perceptible transitions in and health states of overall VA, which would provide a more plausible basis for the transformation to utilities.
The PBAC acknowledged the observations in pre-PBAC response that there may be consequences for quality of life of unilateral blindness that are not captured by overall VA such as depth perception affecting mobility. However, such consequences could only be incorporated in a model with an alternative structure that plausibly linked transitions involving unilateral blindness for such patients to a mapping approach that reflected these consequences in utility terms.
The PBAC also noted that the GALILEO trial excluded patients who had “only one functional eye even if that eye was otherwise eligible for the study”, which means that the other eye of included trial participants functioned well. The EQ-5D results of this trial remained high and similar with a non-significant trend towards a greater difference favouring aflibercept than at baseline. However the PBAC concluded that, as the other eye is not randomly distributed in the study, the utility estimates in the study depend on both eyes, and averaging across the patients in the trial based on visual acuity in the CRVO-affected eye does not capture the relevant effect of treating a unilateral eye condition on overall visual acuity (and therefore utility) for the patients.
The PBAC considered that the lifetime model assumed that, once an eye is treated and vision loss is averted, patients will not suffer a recurrence of macular oedema; or if they do, then treatment will have the same effectiveness. The PBAC noted that there was no clinical trial evidence supporting this assumption, and therefore the PBAC considered it was not a reasonable assumption.
The PBAC acknowledged that the resubmission reduced the excess mortality risk from a hazard ratio of 1.23 (which included direct and indirect effects) in the submission considered in July 2013 to 1.13 (which included direct effects only) (both from Christ et al 2008), however the PBAC re-iterated its previous concerns from July 2013 that the increased mortality risk applied is based on cross-sectional data that does not a show quantifiable causal relationship between visual impairment and death, and is potentially confounded by other factors. Further, the study hazard ratio of 1.13 does not refer specifically to unilateral blindness, but is estimated based on a group of possible visual problems categorised as “some visual impairment”.
The PBAC agreed with the ESC that there may be an increase in mortality risk due to unilateral blindness, but considered the submission’s hazard ratio of 1.13 to be unreliable and likely an overestimate. Based on the evidence provided, the PBAC considered the magnitude of the increase in mortality risk to be unknown. The PBAC noted that when the mortality benefit is removed (RR=1) the ICER increases but remains within the range of $45,000 to $75,000/QALY with OCT costs in the BSC arm and is between $75,000 and $105,000/QALY with no OCT costs in the BSC arm.
The PBAC noted that, while the requested price was decreased compared to the July 2013 price, the ICERs had increased. The model was sensitive to duration, with the ICER increasing to between $75,000 and $105,000/QALY when model duration was limited to 10 years.
The second economic analysis presented in the resubmission was a cost-minimisation analysis based on a claim of non-inferior efficacy and safety between aflibercept and ranibizumab.
The PBAC noted that the equi-effective doses presented in the submission were estimated as an average of 8.6 aflibercept injections over 12 months (weighted from both COPERNICUS and GALILEO trials) and 9.6 injections of ranibizumab injections over 12 months (based on the CRUISE trial).
The PBAC noted that this estimation of equi-effective doses was based on incomplete information: the resubmission only presented the mean re-injection frequency for the CRUISE trial and no information on the distribution of re-injections; both the CRUISE and COPERNICUS trials were affected by cross-over at week 24; and the re-injection frequencies were are only measured up to week 52.
The PBAC also noted that pre-sub-committee response estimated that the average number of injections per patient in the three loading dose regimen would be 6.89. This assumed that the re-injection frequency in the ‘treat and extend’ period is the same following the three loading dose regimen as seen in the clinical trials for the six loading dose regimen, and that the longer term clinical efficacy is not impacted by the change in loading doses.
The PBAC considered that a one to one injection relativity between aflibercept and ranibizumab injections would be more reasonable in the absence of complete data.
The PBAC considered various means by which it might be possible to construct a comparison with compounded bevacizumab, and noted the Department’s advice that, if compounded bevacizumab were to be considered for PBS listing, its pricing for PBS purposes would most likely reflect the approach to pricing already applied to bevacizumab as an oncology medicine through the Efficient Chemotherapy Funding Program. The PBAC noted that it had not assessed the cost-effectiveness of bevacizumab in this setting. This would need to be informed by a comprehensive review of the comparative effectiveness and safety of aflibercept and bevacizumab.
11. Estimated PBS Usage and Financial Implications
The likely number of patients treated per year was estimated in the submission to be less than 10,000 in Year 5, at an estimated net cost per year to the Government of $10 - $30 million in Year 5.
The PBAC noted that the same epidemiological approach was used as in the submission considered in July 2013, with changes made to the uptake rate of aflibercept and the frequency of aflibercept re-injections during the maintenance phase.
The PBAC noted that the estimated costs were highly sensitive to projected uptake rates. The PBAC considered that, if aflibercept were to be PBS-listed as the only PBS-listed treatment for this condition, the submission’s initial Year 1 uptake rate would be a likely underestimate.
The PBAC further noted that the costs are also sensitive to the frequency of injection of aflibercept and that this was likely to be different in clinical practice from that estimated in the resubmission due to the reduction in the number of loading doses in the proposed product information.
The ESC considered that the frequency of repeat aflibercept injections and duration of therapy over an extended time period are difficult to predict, but the duration is likely to be less than that indicated by the utilisation of aflibercept for age-related macular degeneration (AMD). In terms of the frequency of injections, ophthalmology treatment practices may have changed over time, thus the most relevant utilisation patterns might be from patients who have more recently commenced aflibercept.
12. PBAC Outcome
The PBAC rejected the submission on the basis of unacceptably high and likely underestimated cost-effectiveness for aflibercept compared with placebo, and on the basis of inadequate comparative data against compounded bevacizumab.
The PBAC noted that the resubmission had not included a comparison with bevacizumab. The PBAC considered that compounded bevacizumab was also a relevant comparator because evidence of its current use in patients who would be eligible for PBS-subsidised aflibercept means that prescribers would replace compounded bevacizumab in practice with aflibercept if the PBS subsidises aflibercept.
The PBAC noted Departmental advice that, if compounded bevacizumab were to be considered for PBS listing, its pricing for PBS purposes would most likely reflect the approach to pricing already applied to bevacizumab as an oncology medicine through the Efficient Chemotherapy Funding Program.
The PBAC considered that a comprehensive review of the relative effectiveness and safety of aflibercept and bevacizumab would be informative.
The PBAC accepted the claim that aflibercept has superior effectiveness and a similar safety profile to placebo, but remained concerned regarding the long-term efficacy of aflibercept particularly given that the submission did not provide any evidence relating the revised loading dose (three doses given monthly) to longer-term clinical efficacy.
The PBAC considered that an inherent issue in the model structure was the link between BCVA of the CRVO-affected eye (which forms the basis of the transition probabilities used) and utility values which are affected by the overall BCVA of the patient. The PBAC considered that this leads to a significant overestimate of the utility benefits of aflibercept, therefore the resulting incremental cost-effectiveness over placebo is highly favourable to aflibercept.
The PBAC considered that more extensive respecification of the base case would be needed to account for this structural issue given the paucity of data.
Notwithstanding the inherent issue with the model structure in relation to the utility consequences of unilateral blindness being overestimated in the economic model, the PBAC further considered that the lifetime time horizon of the model was not adequately justified given the lack of knowledge of the long-term treatment effect and the potential for recurrence of CRVO.
The PBAC noted that the frequency of repeat injections and duration of therapy of aflibercept in CRVO over an extended period of time are unknown.
The PBAC considered that any resubmission would need to address the issues outlined regarding the model, in particular a more extensive respecification of the base case would be needed.
The PBAC advised that any resubmission would need to be a major resubmission.
Recommendation:
Rejected
13. Context for Decision
The PBAC helps decide whether and, if so, how medicines should be subsidised in Australia. It considers submissions in this context. A PBAC decision not to recommend listing or not to recommend changing a listing does not represent a final PBAC view about the merits of the medicine. A company can resubmit to the PBAC or seek independent review of the PBAC decision.
14. Sponsor’s Comment
Bayer does not agree with PBAC’s consideration that compounded bevacizumab is a relevant comparator in the reimbursement assessment of aflibercept.

